Die EU-HTA-Verordnung im Kontext der EU Triple A:
Access, Availability, Affordability – Zugang, Verfügbarkeit, Erschwinglichkeit
Pharmazeutische Strategie: Implikationen für Österreich
Im Herbst 2020 kündigte die Europäische Kommission ihre (damals) neue „Pharmazeutische Strategie für Europa“ an, die die allzu offensichtlichen Problembereiche „(Europaweiter) Zugang zu innovativen Medikamenten“, „Verfügbarkeit von (insbesondere eingeführten) Medikamenten“ und „Erschwinglichkeit neuer Medikamente“ (A+++: Access, Availability, Affordability) lösen möchte. Eine erste Überarbeitung der bestehenden Rechtsvorschriften erfolgte und wurde im April 2023 vorgelegt.1 Diese wird nun im Europäischen Parlament und im Rat diskutiert. In diesem Kontext wird auch die Schaffung einer „Europäischen Arzneimittelinfrastruktur“ diskutiert, um dem Marktversagen während des gesamten Lebenszyklus eines Arzneimittels (Forschung, Entwicklung, Produktion und Vertrieb) entgegenzutreten.2 Als weiteres Element trat die EU-HTA-Verordnung (HTA Regulation – HTAR) im Jänner 2022 in Kraft,3 die – nach einer Übergangsphase von drei Jahren – ab 2025 rechtsverbindlich umgesetzt sein muss.
Health Technology Assessment (HTA) hat in den letzten zwei Jahrzehnten in allen westlichen Ländern enorm an Bedeutung gewonnen. HTA hat die vergleichende Nutzenbewertung von neu zugelassenen Medikamenten und Medizinprodukten zur Aufgabe und dient zur Entscheidungsunterstützung, welche der medizinischen Interventionen in Leistungskataloge oder Arzneimittellisten aufgenommen werden und solidarisch bezahlt werden sollen. Die EU-HTA-Verordnung regelt in erster Linie die Zusammenarbeit unter europäischen HTA-Institutionen bei der Bewertung von Arzneimitteln und Medizinprodukten. Ziel ist die europaweite Bündelung von HTA-Ressourcen, um qualitativ hochwertige und zeitnahe wissenschaftliche Assessments zu erarbeiten und damit die evidenzbasierte Entscheidungsfindung auf nationaler Ebene zu unterstützen. Dadurch soll ein früherer Zugang zu „echten“ Innovationen erleichtert werden. Neben Effizienzgewinnen in der Erarbeitung von Medikamentenbewertungen sollen aber auch Transparenz und Nachvollziehbarkeit in der Erstellung von Assessments durch Harmonisierung der Methodik auf hohem qualitativem Niveau gewährleistet werden.
In Vorbereitung der HTAR wurden in den letzten Jahren im Rahmen von EUnetHTA (European Network for HTA) Joint Actions 2006–2020 Methodenpapiere, Dokumentvorlagen für Assessments, aber auch für Einreichungen der Industrie erarbeitet, Prozesse und Standard Operating Procedures (SOPs) definiert und nun zuletzt in einem Servicevertrag (EUnetHTA 21) finalisiert, damit die HTAR reibungslos umgesetzt werden kann. Der Vorteil der HTAR für kleine Länder (ohne entsprechende HTA-Ressourcen) ist, dass hochwertige Assessments – zeitnah zur Zulassung durch die European Medicines Agency (EMA) – zur Entscheidungsunterstützung verfügbar werden. In einem mehrjährigen Role-out werden 2025–2028 ausschließlich onkologische Medikamente und Advanced Therapies (ATMP) wie Zell- und Gentherapien gemeinsam bearbeitet (s. Abb.). Ab 2028 sollen dann alle als Orphan Drugs von der EMA zugelassenen Medikamente, aber auch Risikoklasse-III- und -IIb-Medizinprodukte diesem europaweiten Bewertungsprozess unterzogen werden.
Timeline of Implementation
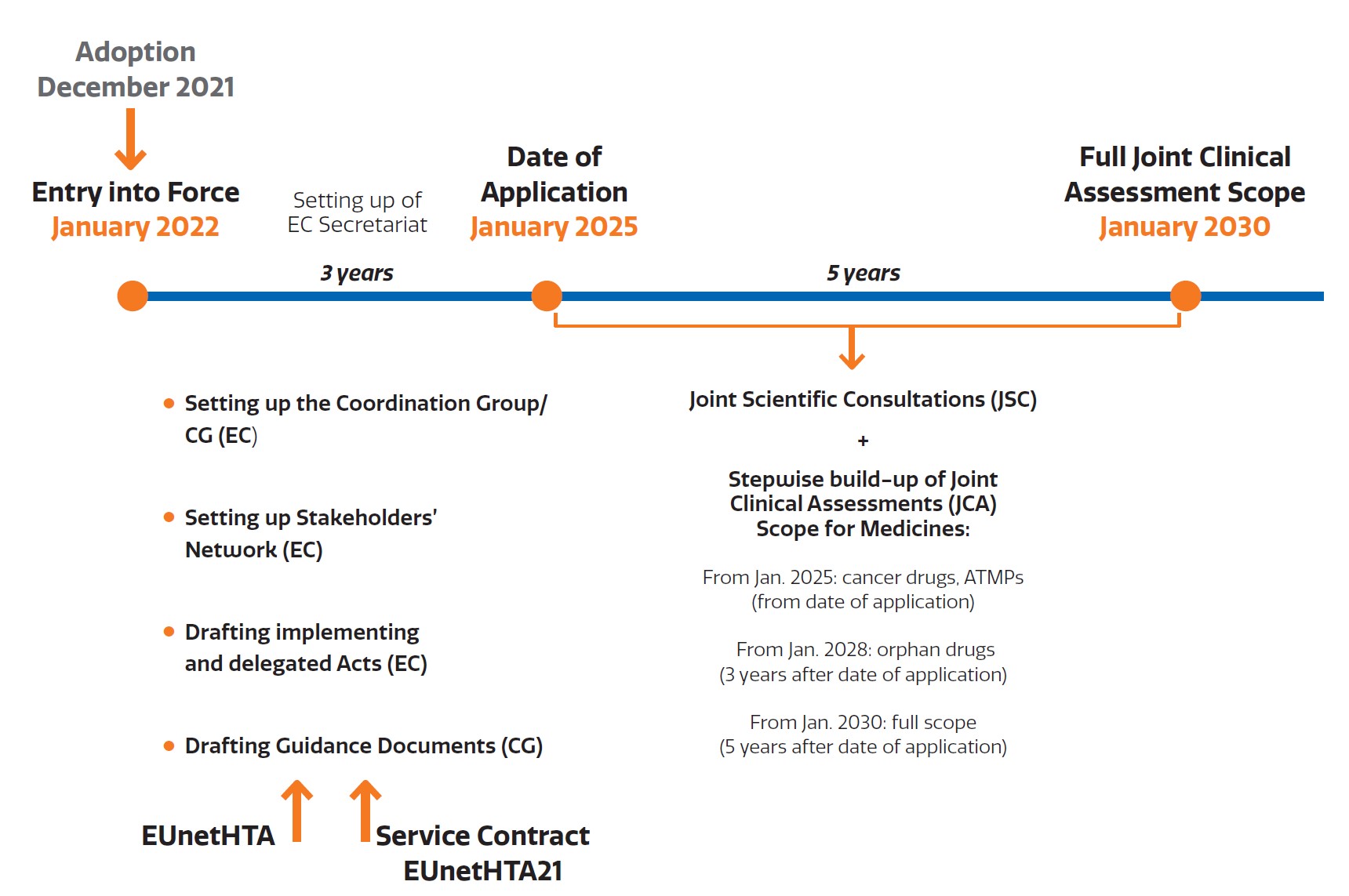
Abb.: Zeitschiene der Implementierung der HTAR
Erwarteter Nutzen der HTAR
Der erwartete Nutzen für die Entscheidungsträger in den EU-Mitgliedsstaaten ist, dass zeitnahe (zeitgleich mit der EMA-Bewertung) wissenschaftliche Berichte zur Unterstützung der evidenzbasierten Entscheidungsfindung auf nationaler Ebene verfügbar werden. Dieser Vorteil wird vor allem von kleinen Ländern wie Österreich, aber auch Belgien, den Niederlanden etc. gesehen. Widerstand kam hingegen – während der Verhandlung der HTAR – von jenen Ländern, die große eigenständige HTA-Institutionen haben (Deutschland, Frankreich) und bei denen eine Umstellung der Prozesse notwendig wird. Der erwartete Nutzen für Patient:innen liegt in der verbesserten Transparenz bei der Erstellung der Bewertungen, aber auch in deren Beteiligung im EU-HTA-Prozess etwa bei der Benennung relevanter klinischer Endpunkte oder relevanter Vergleichstherapien. Ein weiterer Vorteil für Patient:innen soll die verbesserte Verfügbarkeit von Innovationen mit hohem Zusatznutzen auch in Mitgliedsstaaten mit geringerer Kaufkraft durch gegebenenfalls gemeinsame Preisverhandlungen sein. Der erwartete Nutzen für Arzneimittelhersteller sind die einmalige/-zeitige Einreichung der Unterlagen (Dossiers) für die EU-27-Mitgliedsstaaten sowie die klaren und kohärenteren Bewertungsmethoden. Über die Realisierung der Erwartungen an Effizienzgewinne durch geringere Arbeitslast kann derzeit nur spekuliert werden. Da aber ökonomische Bewertungen und systemisch-organisatorische Implikationen (etwa Ort der Verabreichung, Qualifikation von Personal) von den europäischen Bewertungen ausgenommen sind, werden sich die Arbeitsinhalte in nationalen HTA-Institutionen, aber auch in Market-Access-Abteilungen der Arzneimittelhersteller auf diese Aspekte konzentrieren müssen. Die europäischen Assessments sind jedenfalls für die nationale Entscheidungsfindung (verpflichtend) zu verwenden; eine Duplikation, d.h. nationale Assessments zur klinischen Evidenz, ist auszuschließen, Adaptionen an nationale Gegebenheiten wie etwa ökonomische Evaluationen zu Subgruppen und organisatorische Einbettung in klinische Pfade sind jedoch möglich.
Eckpunkte der HTAR
Die europaweite Zusammenarbeit im Bereich HTA steht auf 4 Säulen von Kooperationen:
- Zusammenarbeit beim „Horizon Scanning“ zu „Emerging Technologies“, also der frühen Identifikation von neuen Arzneimitteln, Klassen von Medikamenten und Medizinprodukten, um echte Innovationen frühzeitig zu identifizieren, die Auswirkungen von disruptiven Innovationen zu antizipieren und eine Budgetplanung zu ermöglichen, aber auch, um das Wissen und Daten in Verhandlungen zu nutzen.
- Zusammenarbeit bei der frühen Beratung von Arzneimittelherstellern zu Studiendesigns, relevanten Endpunkten und relevanten Vergleichstherapien, um die für vergleichende Nutzenbewertungen (HTA) notwendigen Studien bereits bei der Planung der Zulassungsevidenz zu berücksichtigen. Derartige „Joint Scientific Consultations“ (JSC) werden bereits seit Jahren parallel zu den EMA-Beratungen durchgeführt.
- Zusammenarbeit bei Arzneimittelbewertungen (Joint Clinical Assessments – JCA), basierend auf stringenten Methoden und transparenten Abläufen, zeitgleich mit dem EMA-Zulassungsprozess und nach Konsultation aller EU-27-Mitgliedsstaaten nach deren Interesse für Informationen zu spezifischen Subpatientenpopulationen oder zu unterschiedlichen Vergleichstherapien. Diese Zusammenarbeit ist seit Jahren erprobt, wenngleich – im Gegensatz zur HTAR – bislang von freiwilliger Natur.
- Die Zusammenarbeit bei der Erstellung von Methodenpapieren ist hingegen ein eher junger Kooperationsbereich, der sehr diskussionsintensiv, aber auch lehrreich ist.
Implikationen für Österreich
Die HTAR greift zwar nicht in nationale Refundierungsentscheidungen ein, setzt aber voraus, dass die Mitgliedsstaaten im zentralen Steuerungsgremium (HTAR Coordination Group) ihre Prioritäten für die Bewertung von Produkten einbringen und dort mit einer Stimme sprechen. In stark dezentral organisierten Ländern wie Österreich, in denen viele Entscheidungen regional fallen, aber auch der Bedarf für gesundheitspolitische Entscheidungen regional entsteht, ist eine österreichweite Koordinierung Voraussetzung für eine erfolgreiche Umsetzung der HTAR. Da bei besonders teuren Therapien (etwa neuropädiatrische Therapien) ein Patiententourismus beobachtet werden konnte, wird schon seit Längerem über die österreichweite Finanzierung ebensolcher Therapien aus einem gemeinsamen „Innovationsfonds“ beraten. Das Ergebnis – ob diese gemeinsame Finanzierung überhaupt beschlossen wird, ob Schwellenwerte je Patient:in oder je Patientenkollektiv für „kostenintensiv“ definiert werden, welche Entscheidungsprozesse aufgesetzt werden – ist offen, jedenfalls aber Voraussetzung für die Umsetzung der HTAR in Österreich: Bei einer gemeinsamen Finanzierung kostenintensiver Therapien in Österreich ...
- ist die frühe Identifikation ebendieser Technologien von Bedeutung,
- müssen österreichweit gültige Anwendungsregeln
(wie sie für die CAR-T-Zell-Therapien erstellt wurden) erarbeitet werden und - müssen die europäischen HTA-Bewertungen dafür verwendet werden.
Zur Erleichterung der Identifikation jener Arzneimittel, die österreichweit geregelt werden sollen, ist Österreich im Herbst 2022 der länderübergreifenden „International Horizon Scanning Initiative“ (IHSI; https://ihsi-health.org) beigetreten, die sich die Förderung von fairen und transparenten Arzneimittelpreisen zur Aufgabe gemacht hat. IHSI wird von 9 europäischen Ländern (Niederlande, Irland, Schweiz, Dänemark, Portugal, Norwegen, Belgien, Schweden, Österreich) finanziert und vom US-amerikanischen Institut ECRI (Emergency Care Research Institute) umgesetzt. Für die Implementierung der HTAR und somit die Verwendung der europäischen JCA sowie deren Einbettung in österreichische Behandlungspfade sind eine stärkere Koordinierung des Informationsbedarfs und die Einrichtung eines Bewertungsboards für die Erstellung der Anwendungsalgorithmen unabdingbar. Eine Koordinierung auf „niedrigster“ Ebene findet durch Vernetzung der entsprechenden Abteilungen der Spitalsträger sowie die Einmeldung und gemeinsame Bearbeitung neuer brisanter Themen (Arzneimittel, Medizinprodukte, IT-/AI-Applikationen) bereits statt.
Claudia Wild
Kommentar
Damit Patient:innen ausschließlich wirksame Krebsmedikamente erhalten, müssen diese zuvor auf Nutzen und potenziellen Schaden untersucht werden. Der Nutzen von neu zugelassenen Medikamenten ist jedoch sehr unterschiedlich: Es gibt Medikamente, die einen bedeutsamen Fortschritt der Medizin bedeuten, und Medikamente, bei denen kein wirklicher Zusatznutzen im Vergleich zu den bereits zugelassenen Medikamenten besteht. Um den „Mehrwert“ eines Medikaments (oder anderer medizinischer Verfahren) zu beurteilen, wird das sogenannte „Health Technology Assessment (HTA)“ angewendet. Unter Health Technology (Gesundheitstechnologien) versteht man alle Interventionen, die Krankheiten verhüten, diagnostizieren oder behandeln sollen.
HTA ist ein transparentes und methodisches Vorgehen, welches Gesundheitstechnologien systematisch und basierend auf wissenschaftlichen Erkenntnissen evaluiert und ihre Effekte auf das Gesundheitswesen einordnet. Dabei wird üblicherweise eine neue Behandlung mit der bzw. den bisherigen Standardbehandlung(en) verglichen und festgestellt, ob sie einen größeren oder zumindest gleichen Nutzen für die Patient:innen hat. Grundlage der Gegenüberstellung können dabei nicht nur der medizinische Nutzen sein, sondern auch wirtschaftliche, soziale und ethische Aspekte. Insbesondere diese Zusammenschau verschiedener Perspektiven soll bei HTA aussagekräftige Ergebnisse erzielen. Von HTA unterstützte Informationen werden für Planungen im Gesundheitswesen herangezogen. Auch Entscheidungen zur Finanzierung von Medikamenten können auf der Grundlage einer solchen Bewertung getroffen werden.
Im Gegensatz zu großen europäischen Ländern, beispielsweise Deutschland, wurde der enorme Aufwand eines HTA in Österreich bisher nicht realisiert. Der Beitrag von Doz.in Dr.in Claudia Wild beschreibt die vorgesehenen Schritte zu einem europaweiten HTA, welches für Krebsmedikamente ab 2025 fix eingeführt wird. Das Ziel der neu implementierten EU-HTA-Verordnung ist ein EU-weit harmonisierter Bewertungsprozess für jedes neue Medikament. Bereits bei Zulassung eines Medikaments soll dieses HTA vorliegen. Das HTA dient allen europäischen Mitgliedsländern als Grundlage für eine nachfolgende Kosten-Nutzen-Bewertung, z.B. eine „Preiseinstufung“ (Kostenersatz) in Österreich.
Armin Gerger
1 Proposal for a regulation laying down union procedures for the authorisation and supervision of medicinal products for human use and establishing rules governing the European Medicines Agency;
2 European Parliamentary Research Service (EPRS) Scientific Foresight Unit (STOA): European Pharmaceutical R&D: Could public infrastructure overcome market failure? PE 697.197, Dec 2021;
3 EU Regulation 2021/2282 of the European Parliament and of the Council of 15 December 2021 on health technology assessment and amending Directive 2011/24/EU;